Технология переработки нефти и газа. Часть 1Пиролиз углеводородов
В настоящее время пиролиз углеводородов является основным источником не только производства олефинов – этилена и пропилена, но и бутадиена, бутиленов, бензола, ксилолов, циклопентадиена, циклопентена, изопрена, стирола, нафталина, нефтеполимерных смол, сырья для производства технического углерода, растворителей, специальных масел.
В бензиновой фракции пиролиза присутствует до 30% (масс.) бензола, 6-7% толуола, 2-2,5 % ксилолов, около 1 % стирола. Фракция С5, содержит до 30% циклопентадиена, включая димеры, и около 10% изопрена. Тяжелая смола (температура кипения >200ºС) имеет в своем составе нафталин и его гомологи, а также небольшое количество тетралина и конденсированных ароматических углеводородов. Кроме того, смола пиролиза содержит некоторое количест во неароматических углеводородов, включая олефины и диены.
Получение ряда химических продуктов из смолы пиролиза успешно конкурирует с традиционными процессами их производства. Так, себестоимость бензола в 1,3 – 1,5 раза ниже, чем в каталитическом риформинге. За счет этого себестоимость этилена также снижается (на 20 – 30 %).
Основным сырьем процесса пиролиза являются этан, пропан, бутаны, содержащиеся в попутных и в нефтезаводских газах, газовые бензины и бензины прямой перегонки нефти, а также рафинат каталитического риформинга, остающийся после удаления ароматических углеводородов из катализата. В последнее время в связи с дефицитом и высокой стоимостью бензиновых фракций в качестве сырья пиролиза применяют также средние и тяжелые нефтяные фракции и даже сырую нефть.
Теоретические основы процесса пиролиза. Производство низших олефинов основано на термическом разложении углеводородного сырья с последующим низкотемпературным разделением полученных продуктов. Все реакции, протекающие при пиролизе, можно разделить на первичные и вторичные.
Основная первичная реакция – разложение исходного углеводорода с образованием водорода, низших алканов, этилена, пропилена и других олефинов. К вторичным – нежелательным реакциям относятся:
дальнейшее разложение образовавшихся олефинов;
гидрирование и дегидрирование олефинов с образованием парафинов, диенов, ацетилена и его производных;
конденсация отдельных молекул с образованием более высокомолекулярных углеводородов, а также более стабильных структур (ароматических углеводородов, циклодиенов и др.).
Все эти реакции при пиролизе протекают одновременно, поэтому особенно важным становится создание таких условий, при которых вторичные реакции были бы сведены к минимуму.
Ненасыщенные углеводороды только при достижении достаточно высокой температуры становятся термодинамически более стабильны, чем соответствующие им парафины. Для этилена, например, эта температура составляет 750ºС.
Сравним термодинамику возможных маршрутов образования олефинов. В первом случае при расщеплении (крекинге) молекулы исходного парафина:
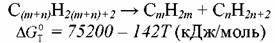 : :
Как известно, термодинамическая стабильность определяется температурой, при которой изменение энергии Гиббса
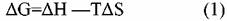
где ΔН — тепловой эффект реакции, Т — температура, ΔS — изменение энтропии,
становится равным нулю или отрицательным.
Обе реакции эндотермичны и протекают с увеличением объема. Чтобы сместить равновесие в сторону расщепления сырья и образования олефинов, необходимо увеличить температуру и снизить давление. Но если крекинг углеводородов происходит с заметной скоростью уже при температуре 500ºС, то вклад реакции дегидрирования в образование продуктов пиролиза становится ощутимым лишь начиная с 800-850ºС. По экономическим соображениям для достижения оптимального парциального давления углеводородов применяют не вакуум, а разбавление исходной смеси водяным паром. Последнее приводит к некоторым как положительным, так и отрицательным последствиям. Положительные связаны:
со снижением удельного количества тепла, необходимого для нагрева труб в реакторе за счет введения части энергии с водяным паром непосредственно в сырье;
улучшением испаряемости углеводородов;
 (не играющим, однако, определяющей роли при температурах до 1000ºС); (не играющим, однако, определяющей роли при температурах до 1000ºС);
со снижением коксоотложения за счет турбулизации потока углеводородов в реакционном змеевике;
с уменьшением вероятности протекания вторичных реакций, вследствие разбавления реакционной смеси.
К отрицательным последствиям относятся затраты энергии на нагрев до температуры реакции, рост инвестиций, связанных с необходимостью увеличения размеров печи и усложнения системы разделения продуктов пиролиза. Необходимое количество вводимого пара зависит в основном от молярной массы исходных углеводородов и лежит в интервале 0,25-1 тонн на тонну сырья соответственно для этана и тяжелых нефтяных фракций.
Итак, основная реакция пиролиза (особенно в случае использования в качестве сырья нефтяных фракций) крекинг углеводородной цепи с образованием олефина и парафина. Ее первичные продукты могут претерпевать дальнейшее расщепление (вторичный крекинг). В конечном итоге получается смесь легких углеводородов, богатая олефинами. Дегидрирование соответствующих олефинов приводит к образованию ацетилена и его производных, а также диеновых углеводородов, обладающих высокой реакционной способностью. Последние в условиях пиролиза вступают в реакции циклизации. При дегидрировании из циклоолефинов получаются арены, в частности бензол, являющиеся, в свою очередь, предшественниками образования полициклических углеводородов и кокса. Протеканию последних реакций (значит и отложению кокса) благоприятствует повышение температуры до 900-1000ºС.
Другой нежелательный процесс полимеризация ненасыщенных углеводородов. Она в условиях пиролиза практически не протекает. Эта реакция экзотермична и начинается лишь при понижении температуры. Быстрое преодоление температурной области, где она уже возможна, и скорость ее еще высока основная задача стадии охлаждения (закалки) газов пиролиза.
Забиваем Сайты В ТОП КУВАЛДОЙ - Уникальные возможности от SeoHammer
Каждая ссылка анализируется по трем пакетам оценки: SEO, Трафик и SMM.
SeoHammer делает продвижение сайта прозрачным и простым занятием.
Ссылки, вечные ссылки, статьи, упоминания, пресс-релизы - используйте по максимуму потенциал SeoHammer для продвижения вашего сайта.
Что умеет делать SeoHammer
— Продвижение в один клик, интеллектуальный подбор запросов, покупка самых лучших ссылок с высокой степенью качества у лучших бирж ссылок.
— Регулярная проверка качества ссылок по более чем 100 показателям и ежедневный пересчет показателей качества проекта.
— Все известные форматы ссылок: арендные ссылки, вечные ссылки, публикации (упоминания, мнения, отзывы, статьи, пресс-релизы).
— SeoHammer покажет, где рост или падение, а также запросы, на которые нужно обратить внимание.
SeoHammer еще предоставляет технологию Буст, она ускоряет продвижение в десятки раз,
а первые результаты появляются уже в течение первых 7 дней.
Зарегистрироваться и Начать продвижение
С увеличением времени пребывания сырья в зоне высоких температур увеличивается вклад нежелательных последовательных превращений целевых продуктов. Поэтому для повышения избирательности (селективности) пиролиза надо уменьшать время контакта. При этом, однако, снижается глубина переработки сырья за проход, а значит, и выход целевых продуктов.
Не только выход продуктов пиролиза, но их состав являются функцией многих параметров, в первую очередь природы сырья и условий осуществления процесса.
Термическое разложение углеводородного сырья происходит по радикальноцепному механизму. Начальное зарождение цепи происходит под влиянием температуры при гомолитическом разрыве наиболее слабой С-С связи с образованием свободных радикалов, которые способны образовывать новый свободный радикал, отрывая атом водорода от молекулы исходного углеводорода.

В общем случае образующиеся первичные радикалы с длинной цепью не устойчивы. Их стабилизация происходит в основном за счет расщепления связи С-С, находящейся в β-положении к радикальному центру, что отвечает общему принципу наименьшего изменения структуры:
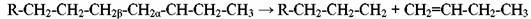
Эта реакция β-распада повторяется до тех пор, пока не образуется сравнительно устойчивый радикал - метильный или этильный, который, в свою очередь, становиться источником зарождения новой цепи. Вероятность образования тех или иных радикалов на стадии продолжения цепи зависит от строения атакуемой молекулы углеводорода. Отрыв атома водорода от третичного атома углерода происходит легче, чем от вторичного и, тем более, первичного атома. В общем случае выход увеличивается с ростом содержания парафинов (нормального строения) в сырье, то есть зависит и от химического состава сырья. Термическая стабильность углеводородов возрастает в ряду парафины<нафтены<арены и уменьшается с ростом длины цепи.
Многообразие протекающих вторичных реакций затрудняет моделирование процесса, особенно при усложнении природы сырья и увеличении степени конверсии. До сих пор при проектировании печей опыт, эмпирические зависимости и экспериментальная проверка играют очень важную роль.
Рассмотрим механизм пиролиза парафинов вначале на примере этана. Инициирование цепи заключается в распаде молекулы С2Н6, по связи С—С на два метильных радикала:

Реакции (1) - (7) описывают образование основных продуктов распада этана на начальных стадиях пиролиза. Согласно имеющимся в литературе данным, основными продуктами пиролиза этана являются этилен и #Н, а радикал •СНЗ образуется в очень малых количествах только на стадии инициирования.
В случае пиролиза пропана на стадии продолжения цепи большую роль играют как радикал #Н, так и метильный радикал СН3.
Инициирование:
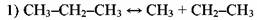
Реинициирование, заключающееся в быстром распаде этильного радикала:
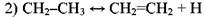
Продолжение цепи, в результате которого могут образоваться изо- или н-пропильный радикалы:
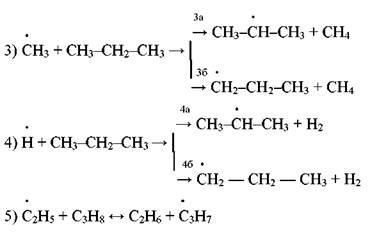
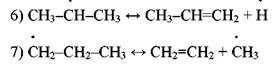
Радикалы Н> и СНЗ* ведут цепь распада дальше. Обрыв цепи протекает так:
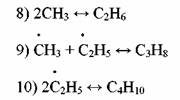
Приведенный механизм распада пропана соответствует составу продуктов только на начальных стадиях процесса.
Большое влияние на состав продуктов пиролиза оказывает температура. При низких температурах, соответствующих процессу крекинга, проявляется большая роль реакций (За) и (4а) по сравнению с (Зб) и (4б), так как энергия разрыва связи С—Н у первичного углеродного атома больше, чем у вторичного. Соответственно образуется больше пропилена по реакции (6) и меньше этилена по реакции (7). Кроме того, при пиролизе углеводородов С4 и выше этильный радикал образуется не только на стадии инициирования, но и на стадии продолжения цепи. В этом случае состав продуктов пиролиза в значительной степени зависит от соотношения скоростей реакций (2) и (5). При низких температурах большую роль играет реакция (5), энергия активации которой составляет около 45 кДж/моль, а роль реакции (2), имеющей энергию активации 168 кДж/моль, значительно меньше. В результате образуется больше этана и меньше этилена. При высоких температурах, напротив, образуется больше этилена, а пропилена и этана меньше. Это объясняется тем, что в соответствии с уравнением Аррениуса, с ростом температуры в большей степени ускоряются реакции, которые имеют более высокую энергию активации, а именно (2), (Зб) и (4б).
Существенное значение для процесса пиролиза имеет стадия обрыва цепи. Порядок реакции по исходному углеводороду зависит от того, которая из трех реакций обрыва цепи (8), (9) или (10) превалирует. Он может составлять 0,5, 1 или 1,5.
Причины торможения распада парафинов олефинами (пропиленом и изобутеном) объясняют двумя механизмами.
Для термического крекинга, где преобладает радикал *CH3 торможение объясняют, так называемым, аллильным механизмом.
Согласно ему радикал *СН3 отрывает атом водорода от пропилена или изобутилена с образованием малоактивного аллильного радикала:
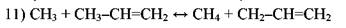
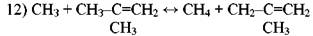
Аллильный радикал не способен продолжать цепь распада, вследствие чего замена радикала *СН3 аллильным приводит к торможению распада парафина. При температурах пиролиза торможение является следствием присоединения водорода к олефину с образованием колебательновозбужденной частицы, которая распадается на этилен и метильный радикал (работы Р. А. Калиненко):
• • • •
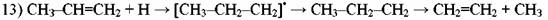
Торможение распада является следствием замены очень активного радикала Н> менее активным радикалом #СНЗ. Установлено также, что парафин ускоряет распад олефина за счет той же реакции (13).
В смесях парафинов с олефином наблюдается предел торможения, который соответствует содержанию 30 - 50% олефина в смеси. Это объясняется тем, что по мере увеличения количества олефина растет концентрация радикалов *СН3, которая компенсирует убыль радикалов Н>. Наиболее сильно торможение проявляется при пиролизе этана, так как цепь ведет радикал Н# (скорость реакции снижается в 7 - 10 раз). Для пропана скорости распада снижаются в 2 -2,5 раза, для н-бутана в 1,2 - 1,3 раза. Это объясняется тем, что при распаде этих углеводородов цепь ведут радикалы Н> и *СН3. Кроме того, все углеводороды, кроме этана, образуют пропилен. Торможение распада пропана и н-бутана является результатом взаимодействия пропилена, как с радикалом Н% так и с радикалом СН3* по реакции (11). Изотопным методом Р. А. Калиненко было установлено, что при высоких температурах (800 - 840ºС) в смесях н-бутана с пропиленом около 60 % пропилена реагирует по реакции (13) и 40 % — по реакции (11). По мере увеличения молярной массы парафина или олефина степень ингибирования снижается и практически перестает сказываться.
Сервис онлайн-записи на собственном Telegram-боте
Попробуйте сервис онлайн-записи VisitTime на основе вашего собственного Telegram-бота:
— Разгрузит мастера, специалиста или компанию;
— Позволит гибко управлять расписанием и загрузкой;
— Разошлет оповещения о новых услугах или акциях;
— Позволит принять оплату на карту/кошелек/счет;
— Позволит записываться на групповые и персональные посещения;
— Поможет получить от клиента отзывы о визите к вам;
— Включает в себя сервис чаевых.
Для новых пользователей первый месяц бесплатно.
Зарегистрироваться в сервисе
Выше были рассмотрены в основном реакции распада, наблюдаемые на начальных стадиях процесса, без учета вторичных реакций и реакций уплотнения, которые играют большую роль в процессе пиролиза. По мере углубления процесса в реакционной смеси появляется все больше продуктов уплотнения и кокса, мешающего нормальному осуществлению процесса. В реакциях уплотнения принимают участие олефины и ароматические углеводороды. В настоящее время отсутствует единая точка зрения на механизм образования высокомолекулярных углеводородов и кокса. Предполагается, в частности, что кокс образуется в результате реакций полимеризации, дегидроциклизации и деструктивной поликонденсации, которые, в конечном счете, ведут к образованию сложных полициклических структур, обедненных водородом:
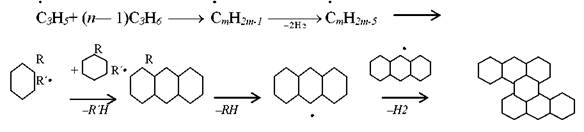
Имеются и другие схемы образования кокса.
Тем не менее, считается что кокс, отлагающийся в реакторе пиролиза, может образовываться двумя путями:
а) гетерогенным разложением молекул углеводородов на стенке реактора или на частицах металла, извлеченных из металлической повер хности и остающихся на поверхности растущего слоя кокса;
б) при реакциях присоединения в объеме реактора, которым особенно благоприятствуют полициклические ароматические углеводороды, содержащиеся в сырье (например, газойлевой фракции).
В пользу представления о двух различных путях образования кокса при пиролизе углеводородов свидетельствует, в частности, разнообразие типов и структур кокса, формирующегося при термическом разложении жидких и газообразных углеводородов. При температурах промышленного пиролиза – от 650 до 900ºС – может формироваться кокс трех типов, нитевидный ленточный (дендрит) или игольчатый, слоистый анизотропный, образующий прочную пленку, и аморфный (“пушистый”), изотропный, образующий относительно непрочную пленку черного цвета.
Количественное соотношение двух путей образования кокса зависит от условий ведения процесса (структура и парциальное давление паров исходных углеводородов, температура реакции, состояние стенок реактора и др.). Кокс, образованный каталитическими реакциями (нитевидный), очевидно, преобладает при относительно низких температурах и на ранних стадиях процесса. При более высоких температурах и значительных степенях превращения исходного сырья, повидимому, возрастает значение конденсационного механизма (получается слоистый анизотропный и аморфный изотропный кокс), причем тип кокса зависит от парциального давления углеводородов, от свойств поверхности, на которой кокс отлагается, строения исходных углеводородов, температуры и ряда других факторов. С увеличением парциального давления углеводородов повышается доля образующегося аморфного кокса.
|

