Технология переработки нефти и газа. Часть 1Термодинамика, кинетика и механизм изомеризации
Реакции изомеризации парафиновых углеводородов являются равновес-ными:
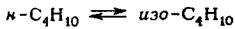
Они протекают практически без изменения объема, поэтому термодина-мическое равновесие зависит только от температуры; низкие температуры бла-гоприятствуют образованию изопарафиновых углеводородов. Тепловой эффект реакции изомеризации невелик - от 2 до 20 кДж/моль - и мало меняется с изме-нением температуры. Исследованию равновесий реакций изомеризации пара-финовых углеводородов посвящено значительное число работ эксперименталь-ного и расчетного характера. Наблюдаемое для некоторых углеводородов не-совпадение объясняется недостаточно точным вычислением термодинамиче-ских величин. При расчете равновесных составов по значениям констант рав-новесия необходимо также учитывать, что на практике при протекании реакции изомеризации не всегда образуются все теоретически возможные изомеры; на-пример, в продуктах изомеризации понтана были обнаружены только два изо-мера - н-пентан и изопентан (2-метилбутан); неопентан (2,2-диметилпропан) не был обнаружен. Последнее вызвано неустойчивостью первичного карбкатиона - необходимой стадии перегруппировки вторичного карбкатиона. Ввиду отсут-ствия неопентана равновесие должно рассматриваться только между н-пентаном и изо-пентаном. То же самое относится к изомерам гептана: при про-ведении изомеризации отсутствуют 2,2-диметилпентан, 3,3-диметилпентан, З-этил- пентан, что связано с затруднениями кинетического характера.
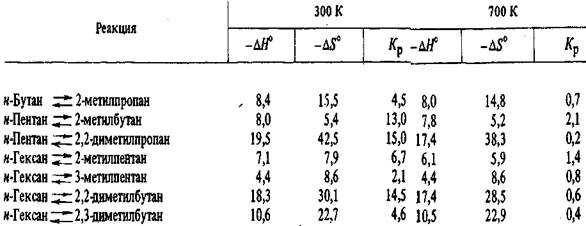
Константы равновесия реакций изомеризации парафинов C4-С6, энталь-пия изомеризации и изменение энтропии изомеризации, вычисленные на осно-вании спектроскопических данных и данных о свободных энергиях, приведены в табл. 5.8, а равновесные составы смесей изомеров - в табл. 5.9. При расчетах констант равновесия реакции изомеризации используется разница в свободных энергиях изомеров:
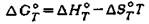
 - энтропия изомеризации. - энтропия изомеризации.
 Дж/ (моль К), и, константы равновесия Кр для реакции изомеризации парафиновых углеводородов С4-С6 в газовой фазе Дж/ (моль К), и, константы равновесия Кр для реакции изомеризации парафиновых углеводородов С4-С6 в газовой фазе
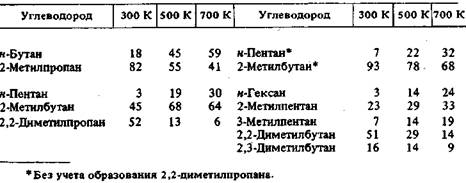
Состав равновесной смеси рассчитывается на основании констант равновесия изомеризации каждого углеводорода в другой:
Таблица 5.9 Массовый состав (в %) равновесных смесей изомеров парафинов С4-С6
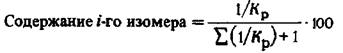
Кинетика и механизм реакции изомеризации зависят от типа катализатора и условий проведения реакции. В условиях гетерогенного катализа реакция изомеризации парафинов протекает по термодинамически контролируемому механизму. Количественной оценкой кинетических параметров реакционной способности углеводородов является константа скорости превращения углево-дорода в изомерный углеводород или смесь изомеров. Изучение путей этих превращений и состава промежуточных продуктов связано с изучением меха-низма реакции.
При осуществлении изомеризации парафиновых углеводородов на про-мышленных алюмоплатиновых катализаторах, промотированных фтором и хлором, металлцеолитных катализаторах, а также сверхкислотах, особенности кинетики и механизма реакции обусловлены механизмом образования проме-жуточных соединений.
Предложено несколько механизмов образования карбкатиона, один из них сводится к межмолекулярному гидридному переносу:
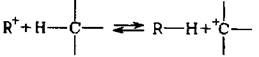
Существует другая точка зрения, согласно которой карбкатионы могут образовываться из парафинов путем отщепления гидрид-иона льюисовской ки-слотой:
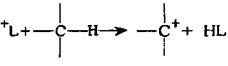
В этом случае путем рекомбинации протона и гидрид-иона может образо-ваться газообразный водород.
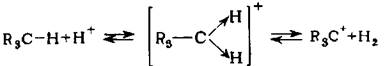
Изомеризация парафиновых углеводородов в сверхкисдотных средах происходит путем протонирования парафинового углеводорода по s -связи, при этом образуется неклассический карбониевый ион с двухэлектронной трехцен-тровой связью, последующее расщепление которого приводит к образованию обычного трехкоординированного карбкатиона и водорода:
Механизм изомеризации на бифункциональных катализаторах. Рассмат-ривая механизм реакции изомеризации парафиновых углеводородов на би-функциональных катализаторах, содержадих металлы VIII группы, можно предположить три типичных случая, в зависимости от кислотности носителя:
на катализаторах с очень сильной кислотностью носителя изомериза-ция происходит на кислотных центрах, роль металла сводится к ограни-чению образования кокса и предохранению от дезактивации кислотных центров; примером может служить процесс на алюмоплатиновом катали-заторе, промотированном хлором;
на катализаторах с очень низкой кислотностью носителя изомеризация происходит только на металлических центрах, и механизм реакции зави-сит от размера кристаллитов металла;
на катализаторах со средней кислотностью носителя, таких как плати-на на аморфном алюмосиликате или на фторированном оксиде алюминия, изомеризация происходит по обычному бифункциональному механизму -образование промежуточных соединений на металлических участках ии-зомеризация олефинов на кислотных участках.
Если изомеризация протекает на поверхности металлов, механизм отли-чается от бифункционального. В случае бифункционального механизма в ад-сорбции парафинового углеводорода на поверхности металла участвуют два соседних атома углерода, от парафинового углеводорода могут отщепляться два атома водорода с образованием олефина в газовой фазе, адсорбированный олефин может подвергнуться гидрогенолизу. Если парафин адсорбируется ато-мами углерода, которые не являются соседними, то возможны образование но-вой С-С-связи, приводящее к пяти- или шестичленным циклическим углеводородам, и их последующее раскрытие за счет разрыва другой С-С-связи. Для протекания такой реакции необходимо, чтобы связанные с поверхностью атомы углерода были разделены четырьмя или пятью атомами углерода и связаны с двумя соседними атомами металла. Возможность протекания такой реакции была открыта Го и Андерсеном. Последовательность реакций при таком меха-низме изомеризации следующая:
Забиваем Сайты В ТОП КУВАЛДОЙ - Уникальные возможности от SeoHammer
Каждая ссылка анализируется по трем пакетам оценки: SEO, Трафик и SMM.
SeoHammer делает продвижение сайта прозрачным и простым занятием.
Ссылки, вечные ссылки, статьи, упоминания, пресс-релизы - используйте по максимуму потенциал SeoHammer для продвижения вашего сайта.
Что умеет делать SeoHammer
— Продвижение в один клик, интеллектуальный подбор запросов, покупка самых лучших ссылок с высокой степенью качества у лучших бирж ссылок.
— Регулярная проверка качества ссылок по более чем 100 показателям и ежедневный пересчет показателей качества проекта.
— Все известные форматы ссылок: арендные ссылки, вечные ссылки, публикации (упоминания, мнения, отзывы, статьи, пресс-релизы).
— SeoHammer покажет, где рост или падение, а также запросы, на которые нужно обратить внимание.
SeoHammer еще предоставляет технологию Буст, она ускоряет продвижение в десятки раз,
а первые результаты появляются уже в течение первых 7 дней.
Зарегистрироваться и Начать продвижение
две несмежные С-Н-связи разрываются, а углеводород адсорбируется на поверхности атомами углерода, связанными с соседними центрами ме-талла;
связь С -С образуется между двумя адсорбированными атомами угле-рода, что приводит к образованию циклопентанового или циклогексано-вого кольца, которые могут десорбироваться;
циклические частицы могут повторно адсорбироваться на поверхности или замешать атомы углерода, которые связаны с поверхностью, на дру-гие без десорбции, а С-С-связь может разорваться;
4) присоединение атома водорода к адсорбированным частицам и десорбция без образования С-С-связи приводят к скелетной изомеризации н-гексана в 2-метилпентан.
На рис. 5.16 показана схема изомеризации для парафиновых углеводородов С6 на поверхности металла.
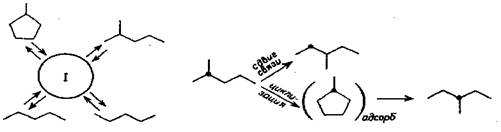
Рис.5.16 Схема реакции изомеризации гексана на поверхности металла: I - пя-
титичленное кольцо на поверхности.
Рис. 5.17 Сдвиг связи и циклическая изомеризация 2-метиллентана - 2-13C. вы-сказаны соображения о комбинировании бифункционального и кислотного ме-ханизмов на платиноморденитсодержащем катализаторе.
Вторым механизмом изомеризации парафинов на металлах является механизм сдвига связи. Он предусматривает образование а-, a-,g -триадсорбированных соединений, связанных с двумя соседними атомами металла. Изомеризация не-опентана должна включать адсорбцию на двух атомах металла, образование связи между атомами углерода на поверхности и разрыв одной из связей в ко-роткоживущем циклопропановом кольце с образованием 2-метилбутана.
Изучение реакции изомеризации гексанов с помощью меченых атомов 13С позволило определить соотношение механизмов реакции сдвига связи и циклической изомеризации в зависимости от свойств катализатора. Оценка размеров кристаллитов платины в катализаторе показала, что в случае кристал-литов размером менее 2 нм преобладают циклическая изомеризация и неселек-тивный гидрогенолиз метилциклопентана, в то время как на более крупных кристаллитах преобладают сдвиг связи и селективный гидрогенолиз.
Для случая металлцеолитных катализаторов не существует однозначной точки зрения на механизм реакции изомеризации парафиновых углеводородов: ряд авторов высказывается в пользу бифункционального механизма, для мор-денитсодержащего катализатора существует предположение о чисто кислотном механизме; высказаны соображения о комбинировании бифункционального и кислотного механизмов на платиноморденитном катализаторе.
Относительная сила металлических и кислотных центров определяет ли-митирующую стадию реакции.
|

