Технология переработки нефти и газа. Часть 1Кинетика изомеризации парафиновых углеводородов
Во всех работах, посвященных кинетике изомеризации парафиновых углеводородов на би-функциональных катализаторах, за исключением, стадией, лимитирующей об-щую скорость реакции изомеризации, считается алкильная перегруппировка карбкатионов. Эта точка зрения подтверждается данными о селективном дейст-вии различных промоторов и ядов на металлические и кислотные участки ката-лизатора. Серии опытов по влиянию фтора, натрия, железа и платины на актив-ность алюмоплатиновых катализаторов в реакции изомеризации н-гексана про-водились при 400oС, давлении 4 МПа и изменении объемной скорости подачи н-гексана от 1,0 до 4,0 ч-1. Опыты на платинированном оксиде алюминия, про-мотированном различными количествами фтора - от 0 до 15% (рис. 4.18), пока-зали, что по мере увеличения количества фтора в катализаторе до 5% наблю-дался значительный рост его изомеризующей активности; поскольку удельная поверхность катализатора не подвергалась заметным изменениям, рост катали-тической активности объясняется изменением химических свойств активной поверхности, а именно усилением кислотности.
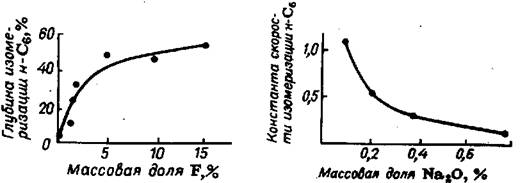
Рис. 5.18 Влияние содержания фтора в катализаторе на глубину изомери-зации н-гексана.
Рис. 5.19 Влияние содержания натрия в катализаторе на скорость изоме-ризации н-гексана.
В опытах с платинированным алюмосиликатом изменение исходной изо-меризующей активности катализатора достигалось отравлением катализатора натрием (рис. 5.19). При увеличении количества Na2O от 0,09 до 0,8%, т. е. в 9 раз, константа скорости изомеризации уменьшалась примерно тоже в 9 раз.
Таким образом, условием получения высокоактивного платинового катализатора изомеризации является применение носителя, обладающего высокой кислотностью.
Далее было исследовано влияние примесей в носителе, которые, не вызывая существенного изменения его кислотности, являются ядами в реакциях гидрирования и дегидрирования.
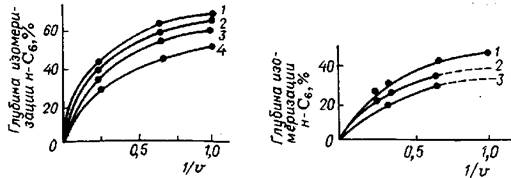
Рис. 5.20. Влияние железа на активность катализатора: Кривая [Fe2O3], % k ; 1 -0,05-1,1; 2 - 0,1-1,0; 3 - 0,2-0,8; 4 - 0.45-0,75
Рис. 5.21. Влияние платины на активность катализатора: Кривая [Рt],% k , 1 – 0,34-1; 2 - 0,1-0.22; 3 - 0,16-0,025
Образцы платинированного алюмосиликата, в которые вводились раз-личные количества железа (в виде Fe2O3), были испытаны в реакции изомери-зации н-гексана (рис. 5.20). Активность катализатора при увеличении содержа-ния Fe2O3 в 15 раз снижалась лишь в 2,4 раза. То обстоятельство, что резкое из-менение дегидрирующей активности катализатора обуславливает лишь относи-тельно небольшое уменьшение глубины изомеризации, подтверждается опыта-ми, проведенными на образцах катализатора, в которых массовая доля платины изменялась от 0,025 до 1%,. т. е. в 40 раз. При этом константа скорости реакции изомеризации н-гексана возросла лишь в два раза (рис. 5.21).
Общность кинетических закономерностей для различных катализаторов (на всех катализаторах наблюдается первый порядок реакции по углеводороду и торможение реакции избытком водорода) также указывает на то, что лимити-рующей является стадия, протекающая на кислотных центрах носителя.
В предельных случаях низкого и высокого парциального давления водо-рода и пониженного содержания металла лимитирующими могут стать стадии, происходящие на металлических центрах катализатора.
В случае металлцеолитных катализаторов лимитирующая стадия зависит от общей поверхности платины и размера кристаллитов металла; для катализа-торов с большой поверхностью металла лимитирующая стадия - скелетная изо-меризация олефинов.
Кинетические закономерности изомеризации н-пентана на промыш-ленных катализаторах. Представляет интерес анализ кинетических законо-мерностей, полученных при изучении изомеризации парафиновых углеводоро-дов, например н-пентана, на промышленных катализаторах Pt -А12O3-F, Pt-А12O3-С1 и Рt - HМ, особенно если учесть, что механизм протекания данной реакции на них различен. Изучение кинетики реакции проводилось в проточной установке при циркуляции водородсодержащего газа.
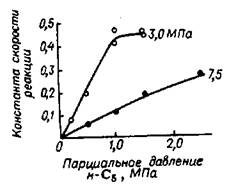
Рис. 5.22 Влияние парциального давления н-пентана на скорость реакции изомеризации. Циф-ры на кривых - парциальное давление водорода.
Из данных рис. 5.22 видно, что при парциальном давлении водорода 3,0 МПа скорость реакции растет пропорционально увеличению парциаль-ного давления пентана до 1,0 МПа; дальнейшее увеличение его до 1,5 МПа не влияет на скорость реакции, подтверждением чему служит практическое постоянство константы скорости реакции при изменении парциального давления н-пентана в этих пре-делах.
При парциальном давлении водорода 7,5 МПа скорость реакции возрас-тала пропорционально увеличению парциального давления н-пентана вплоть до 2,5 МПа.
Различный ход кривых на рис. 5.223 объясняется тем, что при парциаль-ном давлении водорода 3,0 МПа насыщение поверхности катализатора н-пентаном наступает при парциальном давлении последнего, близком к 1,0 МПа; при увеличении парциального давления водорода до 7,5 МПа этот предел на-сыщения сдвигается в область парциальных давлений н-пентана, превышаю-щих 2,5 МПа.
Наблюдаемые факты указывают на существование адсорбционного рав-новесия между реагирующими веществами на поверхности платинового катализатора, которое в случае повышения парциального давления водорода сдви-гается в сторону преимущественной адсорбции водорода и вытеснения н-пентана с поверхности катализатора. Действительно, как указывают литератур-ные данные, относительные адсорбционные коэффициенты для н-пентана и во-дорода при 400oС на платиновом катализаторе равны соответственно 1,0 и 6,4. Из сделанного наблюдения следует практический вывод: при осуществлении процессов при общем рабочем давлении 4,0 МПа целесообразно ограничить парциальное давление м-пентана величиной, не превышающей 1,0 МПа.
Результаты опытов по влиянию парциального давления водорода на про-цесс изомеризации н-пентана при парциальных давлениях н-пентана 0,5 и 1,0 МПа и изменении парциального давления водорода от 1,5 до 9,0 МПа даны на рис. 5.23. При парциальном давлении н-пептона 0,5 МПа и температуре 360, 380 и 400oС уменьшение парциального давления водорода в пределах от 7,5 до 1,5 МПа приводит к пропорциональному увеличению константы скорости ре-акции изомеризации. При парциальном давлении н-пентана 1,0 МПа, темпера-туре 380oС и изменении парциального давления водорода в тех же пределах на-блюдались несколько иные закономерности. Уменьшение парциального давле-ния водорода от 9,0 до 6,0 МПа вызывало пропорциональное снижение кон-станты скорости реакции, однако далее эта зависимость нарушалась.
Забиваем Сайты В ТОП КУВАЛДОЙ - Уникальные возможности от SeoHammer
Каждая ссылка анализируется по трем пакетам оценки: SEO, Трафик и SMM.
SeoHammer делает продвижение сайта прозрачным и простым занятием.
Ссылки, вечные ссылки, статьи, упоминания, пресс-релизы - используйте по максимуму потенциал SeoHammer для продвижения вашего сайта.
Что умеет делать SeoHammer
— Продвижение в один клик, интеллектуальный подбор запросов, покупка самых лучших ссылок с высокой степенью качества у лучших бирж ссылок.
— Регулярная проверка качества ссылок по более чем 100 показателям и ежедневный пересчет показателей качества проекта.
— Все известные форматы ссылок: арендные ссылки, вечные ссылки, публикации (упоминания, мнения, отзывы, статьи, пресс-релизы).
— SeoHammer покажет, где рост или падение, а также запросы, на которые нужно обратить внимание.
SeoHammer еще предоставляет технологию Буст, она ускоряет продвижение в десятки раз,
а первые результаты появляются уже в течение первых 7 дней.
Зарегистрироваться и Начать продвижение
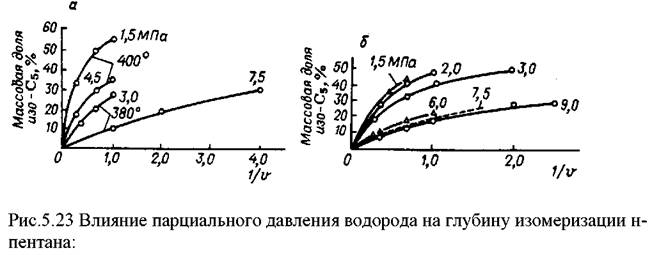
а - парциальное давление н-пептона 0,5 МПа, б - 1,0 МПа. Цифры на кривых -парциальное давление водорода.
Как видим, при изменении парциального давления водорода от 6,0 до 3,0 МПа, т. е. в 2 раза, константа скорости реакции изменялась в 3 раза.
Как уже указывалось, при осуществлении опытов при парциальном дав-лении водорода 3,0 и 7,5 МПа изменение константы скорости реакции изомери-зации с увеличением парциального давления н-пентана носит разный характер. При парциальном давлении водорода 3,0 МПа и н-нептена 1,0 МПа наступает перегиб кривой; далее кривая идет параллельно оси абсцисс. В связи с этим ки-нетические закономерности при парциальном давлении н-пентана 1,0 МПа
справедливы только в области высоких рабочих давлений (6,0-9,0 МПа), когда еще не наступает насыщение поверхности катализатора н-пентаном.
На основании результатов, полученных при изучении влияния парциаль-ного давления водорода и н-пентана, можно предположить, что изменение ра-бочего давления в пределах 1,5-10,0 МПа не должно отражаться на скорости реакции при условии сохранения парциального давления н-пентана не выше 1,0 МПа. При дальнейшем повышении парциального давления н-пентана увеличе-ние рабочего давления должно приводить к торможению реакции изомеризации.
Что касается самого факта торможения реакции изомеризации н-пентана водородом, то в соответствии с установившимся в настоящее время взглядом на механизм реакции изомеризации н-парафиновых углеводородов на бифункцио-нальных катализаторах, реакция протекает через стадию дегидрирования н-алкана с образованием олефинового углеводорода. Следуя этой схеме, тормо-жение реакции водородом можно объяснить снижением концентрации олефина вследствие гидрирования его в парафиновый углеводород, а также явлениями адсорбционного вытеснения пентана водородом с поверхности катализатора.
|

