Технология переработки нефти и газа. Часть 1Реакции, сопровождающие изомеризацию
Реакция изомеризации сопровождается рядом побочных реакций - крекинга, гидрокрекинга и диспро-порционирования; так, молекула н-гексана может подвергаться гидрокрекингу в следуюшие продукты:
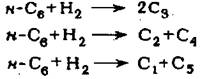
Необходимо отметить, что основным направлением реакций гидрокре-кинга является превращение н-гексана в пропан и бутан, реакция с образовани-ем метана практически не имеет места. Реакция диспропорционирования про-исходит с образованием парафиновых углеводородов с более низкой и высокой молекулярной массой:

При протекании реакции диспропорционирования парафиновых углево-дородов на морденитсодержащих цеолитных катализаторах в продуктах реакции не обнаруживаются углеводороды с молекулярной массой выше исходного, так как имеет место реакция их гидрокрекинга

Поэтому основная реакция при осуществлении диспропорционирования гексанов на цеолитном катализаторе в среде водорода приближенно соответст-вует суммарному уравнению
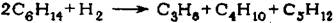
О механизме реакции диспропорционирования парафиновых углеводоро-дов существуют различные точки зрения: для алюмоплатиновых катализаторов, модифицированных хлором, предполагают следующее протекание реакций :
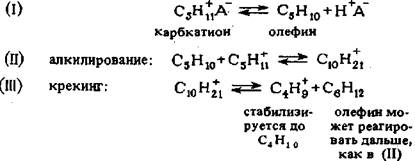
Карбкатион в кислотной системе находится в равновесии с олефинами (I), которые совместно с карбкатионом могут подвергаться алкилированию (II) и реакции крекинга (III). Гексен, образующийся по уравнению (III), может алки-лироваться с С5-карбкатионом до С11 -карбкатиона (по схеме 11), последний подвергается крекингу и т. д. В случае гексанов и гептанов реакции диспропор-ционирования могут протекать значительно быстрее, так как более высокомо-лекулярные карбкатионы могут присутствовать в более высоких концентраци-ях. Результатом высокой концентрации высших олефинов будет гидридный пе-ренос от олефина к карбкатиону, что приведет к образованию аллильных ионов карбония и в свою очередь, к дезактивации катализатора. Реакцию диспропор-ционирования можно подавить, снизив концентрацию карбкатиона и, следова-тельно, концентрацию олефинов. Добавление водорода может сдвинуть равно-весие по уравнению

Уменьшение концентрации карбкатиона приводит к снижению скорости диспропорционирования. Существует мнение, что на цеолитсодержащих ката-лизаторах, в частности на мордените в катионзамешенных формах (Н, Мо, Са, Sr), диспропорционирование протекает за счет реакций трансметилирования. По некоторым данным, наиболее существенную роль играют отщепление и масс-межмолекулярный перенос С2-фрагментов. Ряд исследователей высказы-ваются за механизм реакций по схеме конденсация - крекинг, т. е. без образова-ния низкомолекулярных алкилирующих агентов.
В промышленных процессах изомеризации н-пентана и н-гексана на ка-тализаторах Pt-А12O3-F, Pt-А12O3-CI, Pt-HM-А12O3 найден баланс между реак-циями изомеризации, гидрокрекинга и диспропорционирования, который по-зволяет осуществить процесс с высокой селективностью; в случае изомериза-ции гептанов не достигнуто удовлетворительных результатов.
При осуществлении процессов изомеризации пентан-гексановых фрак-ций, выкипающих до 70oС, в состав их помимо пентанов и гексанов входят вы-сококипящие парафиновые углеводороды, нафтеновые и ароматические угле-водороды (бензол, метилциклопентан, циклогексан, гептаны).
В процессе изомеризации на платиновых катализаторах эти углеводороды подвергаются превращениям в соответствии с условиями термодинамического равновесия для каждого углеводорода по нижеследующим реакциям:
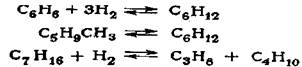
Влияние нафтенов на активность катализаторов в реакции изомеризации парафинов различается в зависимости от природы катализатора и условий осу-ществления реакции. В процессе высокотемпературной изомеризации на алю-моплатиновом катализаторе, промотированном фтором, нафтены, пока их мас-совая доля не превышает 15%, практически не оказывают влияния на глубину изомеризации парафинового углеводорода.
В процессе низкотемпературной изомеризации на алюмоплатиновом ка-тализаторе, промотированном хлором, в присутствии нафтенов скорость реак-ции снижается. В случае осуществления реакции изомеризации н-гексана в жидкой фазе на сверхкислотных катализаторах влияние нафтеновых углеводо-родов специфично. Как следует из рассмотрения результатов, представленных на рис. 5.24, циклогексан оказывает промотирующее действие на скорость изо-меризации н-гексана на HF-SbF5; в противоположность этому реакции крекинга и диспропорционирования подавляются.
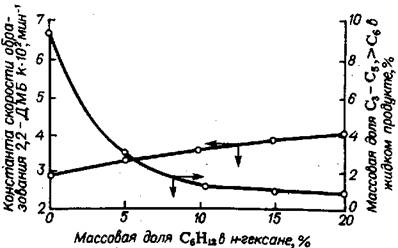
Рис. 5.24 Влияние циклогексана на константу скорости образования 2,2-диметил-бутана и образование побочных продуктов при изомеризации н-гексана: Т=50°C; РН2 =0,4МПа; мольноеотношение С6Н14: MF5=10, HF:MF5=10; время реакции 30 мин.
Влияние циклогексана на скорость реакции изомеризации н-парафина и побочных реакций уменьшается по мере увеличения содержания циклогексана в исходном сырье. Для объяснения полученных результатов было изучено пре-вращение циклогексана в присутствии HF - SbF5, которое показало, что помимо изомеризации циклогексана в метилциклопентан наблюдается образование изо-гексанов в количествах до 8% (рис. 5.25).
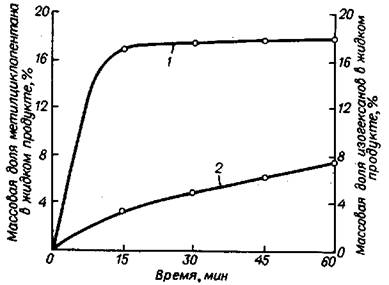
Рис. 5.25 Кривые накопления метилциклопентана (1) и изогексанов (2) при изомеризации циклогексана: Т=50°C; РН2 =0.4 MПa; мольное отношение С6Н12:MF5=15, HF:MF5=10.
На основании полученных результатов превращение циклогексана в при-сутствии HF - SbF5 можно представить следующим образом: при протолизе С-
С-связи циклогексана образуется циклогексониевый ион, который, расщепля-ясь, дает гексильный катион:
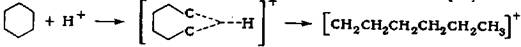
В результате этой реакции катализатор переходит в R-форму - раствор C6H13SbF6- в HF. Следующим актом является образование циклогексильного иона, который претерпевает перегруппировку в метилциклопентильный катион и отрывает гидрид-ион от нейтральной молекулы углеводорода:
Забиваем Сайты В ТОП КУВАЛДОЙ - Уникальные возможности от SeoHammer
Каждая ссылка анализируется по трем пакетам оценки: SEO, Трафик и SMM.
SeoHammer делает продвижение сайта прозрачным и простым занятием.
Ссылки, вечные ссылки, статьи, упоминания, пресс-релизы - используйте по максимуму потенциал SeoHammer для продвижения вашего сайта.
Что умеет делать SeoHammer
— Продвижение в один клик, интеллектуальный подбор запросов, покупка самых лучших ссылок с высокой степенью качества у лучших бирж ссылок.
— Регулярная проверка качества ссылок по более чем 100 показателям и ежедневный пересчет показателей качества проекта.
— Все известные форматы ссылок: арендные ссылки, вечные ссылки, публикации (упоминания, мнения, отзывы, статьи, пресс-релизы).
— SeoHammer покажет, где рост или падение, а также запросы, на которые нужно обратить внимание.
SeoHammer еще предоставляет технологию Буст, она ускоряет продвижение в десятки раз,
а первые результаты появляются уже в течение первых 7 дней.
Зарегистрироваться и Начать продвижение
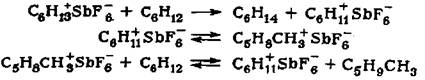
Из представленной схемы видно, что изомеризация циклогексана сопро-вождается образованием изогексанов. Реакция протекает до тех пор, пока все гексильные катионы в комплексе R+MF6- не будут замещены на циклогексиль-ный или метилодклопентильный ион С6Н11+, что хорошо согласуется с экспе-риментальными результатами. Водород не участвует в образовании гексанов, скорость реакции не изменяется в отсутствие водорода (табл. 3).
Таблица 5.10 Превращение циклогексана на сверхкислотном катализаторе HF-SbF5 Условия: температура 50oС; мольное отношение С6Н12:MF5=15, HF:MF5=10; время контакта 60 мин.

Отсюда можно сделать вывод, что влияние нафтенов на превращение па-рафиновых углеводородов определяется характером взаимодействия нафтенов с катализатором. В сверхкислотных средах HF - SbF5 нафтены образуют ком-плекс RMF6, как и парафиновые углеводороды. Скорость образования этого комплекса для различных углеводородов неодинакова и убывает в ряду С6Н12 ≈
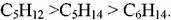 Эта закономерность объясняет, почему при добавлении циклогексана скорость изомеризации н-гексана увеличивается, а для н-пентана остается неизменной. Эта закономерность объясняет, почему при добавлении циклогексана скорость изомеризации н-гексана увеличивается, а для н-пентана остается неизменной.
Нафтеновые углеводороды являются более сильными органическими ос-нованиями, чем парафины, поэтому присутствие их в сырье должно снижать возможность прямого присоединения карбкатиона по s -связи парафинов, кото-рое ведет к образованию продуктов распада и циклических углеводородов.
Можно предположить также, что образование продуктов крекинга и дис-пропорционирования протекает по механизму, включающему участие олефи-нов; в этом случае роль нафтенов должна сводиться к алкилированию непредельных соединений.
На алюмоплатиновых катализаторах, промотированных фтором, взаимного влияния пентанов и гексанов при изомеризации не наблюдается. В случае проведения реакции на алюмоплатиновом катализаторе, промотированном хлором, и на морденитсодержащих катализаторах с увеличением содержания пентанов в сырье увеличивается выход изогексанов, в том числе и 2,2-диметилбутана.
Показано, что при смешении н-пентана и н-гексана скорость изомеризации н-пентана снижается, в то время как скорость изомеризации н-гексана уве-личивается (рис. 5.26). Эти результаты соответствуют гипотезе о том, что н-гексан адсорбируется сильнее, чем н-пентан. Так как молекулы н-пентана и н-гексана достаточно малы, чтобы не задерживаться в порах морденита, то диффузия в порах не должна оказывать влияние на константу скорости реакции. Следовательно, единственное объяснение наблюдаемого явления - это преимущественная адсорбция н-гексана на активных центрах морденита. Статистический анализ показывает, что модель, соответствующая преимущественной адсорбции н-гексана, более корректна.
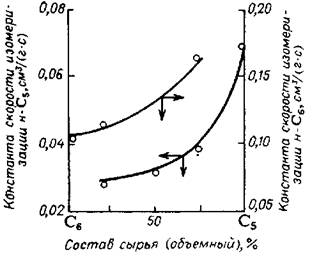
Рис. 5.26. Изомеризация н-пентана, н-гексана и их смесей на Pt-НM - А12О3: Т=140°С, РН2 =2,2 МПа: мольное отношение Н2 : углеводород = 10.
Приведенные здесь данные имеют большое практическое значение. При выборе состава сырья, оценке результатов и показателей изомеризации парафи-новых углеводородов в различных процессах необходимо учитывать углеводо-родный состав сырья, в особенности содержание пентанов и нафтенов; увеличение содержания пентанов всегда приводит к более благоприятному протека-нию процесса изомеризации, в частности к более высокому октановому числу получаемого изомеризата.
Вопрос о влиянии водорода на протекание реакции изомеризации пара-финовых углеводородов на рассмотренных катализаторах обсуждался в ряде работ, но до сих пор не получил однозначного толкования .
На алюмоплатиновом катализаторе, промотированном фтором, реакция изомеризации парафиновых углеводородов не происходит в отсутствие водорода; если катализатор модифицирован хлором, реакция в начальный период протекает и в отсутствие водорода (то же явление имеет место и на фторидах металлов V и VI групп, активированных фтороводородом), но с течением вре-мени ее скорость постепенно уменьшается.
Чтобы реакция изомеризации на металлсодержащем катализаторе проте-кала постоянно, ее необходимо осуществлять в среде водорода. Это связано с явлениями адсорбции и диссоциации водорода на металле и переноса частиц водорода с металла на носитель. Имеют место также явления конкурентной ад-сорбции водорода и промежуточных ненасыщенных соединений на поверхно-сти катализатора, при этом часть этих соединений вытесняется водородом с по-верхности катализатора, что также обеспечивает его стабильную работу.
Несмотря на различный механизм превращения парафиновых углеводо-родов на всех рассмотренных катализаторах, для них наблюдается общность кинетических закономерностей н торможение реакции изомеризации парафи-новых углеводородов избытком водорода. Для всех катализаторов зависимость скорости реакции от парциального давления водорода носит экстремальный характер после достижения определенной концентрации водорода на поверхности катализатора. Величина и положение максимума зависят от типа катализатора, температуры и молекулярной массы парафинового углеводорода.
При низком давлении водорода скорость реакции изомеризации опреде-ляется скоростью образования промежуточных ненасыщенных соединений, ко-торые десорбируются в газовую фазу путем вытеснения их с поверхности ката-лизатора водородом. Таким образом, возрастание скорости реакции изомериза-ции при увеличении парциального давления водорода от нуля до определенной величины связано с явлениями ограничения избыточных концентраций проме-жуточных ненасыщенных соединений; тем самым водород препятствует обра-зованию из них прочно адсорбированных соединений на поверхности катализа-тора. С увеличением парциального давления водорода выше определенного промежуточные соединения и водород начинают конкурировать за участки по-верхности, ответственные за .протекание реакции, и дальнейшее увеличение давления водорода приводит к уменьшению скорости реакции.
В случае осуществления реакции на алюмоплатиновых катализаторах, промотированньах фтором и хлором, и на металлцеолитных катализаторах ско-рости реакций гидрокрекинга и диспропорционирования имеют максимальное значение в отсутствие водорода, постепенно уменьшаются при увеличении парциального давления водорода до некоторого предела и увеличиваются при дальнейшем его повышении.
Сервис онлайн-записи на собственном Telegram-боте
Попробуйте сервис онлайн-записи VisitTime на основе вашего собственного Telegram-бота:
— Разгрузит мастера, специалиста или компанию;
— Позволит гибко управлять расписанием и загрузкой;
— Разошлет оповещения о новых услугах или акциях;
— Позволит принять оплату на карту/кошелек/счет;
— Позволит записываться на групповые и персональные посещения;
— Поможет получить от клиента отзывы о визите к вам;
— Включает в себя сервис чаевых.
Для новых пользователей первый месяц бесплатно.
Зарегистрироваться в сервисе
Итак, существует оптимальное соотношение концентрации водорода и углеводорода на поверхности катализатора, при котором устанавливается рав-новесие между процессами регенерации поверхности катализатора водородом и адсорбционным вытеснением молекул углеводорода водородом с поверхности катализатора и ограничением протекания побочных. реакций. Определение об-ласти оптимального соотношения очень важно для выбора технологических па-раметров процесса, определяющих активность, селективность и стабильность катализатора. Было показано, что в случае осуществления реакции изомериза-ции н-гексана на HF-SbF5 с увеличением парциального давления водорода ско-рость реакций гидрокрекинга и диспропорционирования н-гексана снижается, одновременно несколько снижается и скорость его изомеризации (рис.5.27).
Рис. 5.27. Зависимость константы скорости образования 2,2-диметилбутана (I) и выхода побочных продуктов (2) от парциального давления водорода при изо-меризации н-гексана: Т=50oС; мольное отношение С6Н14- SbF5 =10:1; HF: SbF5 =10: 1; время реакции 60 мин.
Согласно взглядам Ола, взаимодействие карбкатиона и молекулы пара-финового углеводорода протекает через образование промежуточного пентако-
В присутствии катализаторов НС1 - А1С13 и HF - ВF3 ингибирование во-дородом реакции гидрокрекинга и диспропорционирования, а также реакции изомеризации объясняется снижением концентрации карбкатионов по реакции
 ординированного иона, который может расщепляться по нескольким вариан-там: ординированного иона, который может расщепляться по нескольким вариан-там:
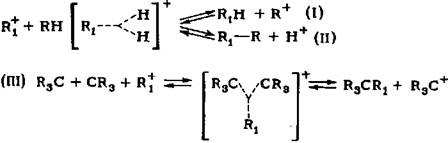
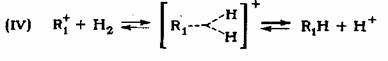 карбкатион вступает еще в одну реакцию: карбкатион вступает еще в одну реакцию:
Эта реакция конкурирует с реакциями (I) - (III). Таким образом, водород снижает скорости реакций изомеризации, гидрокрекинга и диспропорциониро-вания парафиновых углеводородов в присутствии сверхкислот. Как уже упоми-налось, высказано предположение об участии водорода в медленной стадии реакции изомеризации адсорбированных фрагментов на кислотных центрах ката-лизатора.
|

